Kinematic Self-Replicating Machines
© 2004 Robert A. Freitas Jr. and Ralph C. Merkle. All Rights Reserved.
Robert A. Freitas Jr., Ralph C. Merkle, Kinematic Self-Replicating Machines, Landes Bioscience, Georgetown, TX, 2004.
4.3.1 Prions
Prions have been described by Prusiner [1733] as “the only known example of infectious pathogens that are devoid of nucleic acid. All other infectious agents have genomes composed of either RNA or DNA that direct the synthesis of their progeny.” In mammals, prions (Figure 4.12) replicate by recruiting normal cellular prion protein and stimulating its conversion to the disease-causing prion isoform (e.g., “scrapie prion”) [1733]. Modified (pathogenic) prion protein acts as a template upon which normal prion protein is refolded into a nascent modified protein through a process facilitated by another protein [1734].
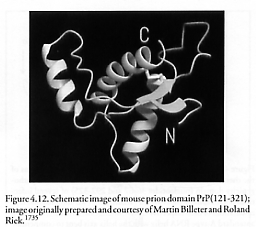
The normal cellular prion protein is converted into modified protein through a post-translational process during which it acquires a high beta-sheet content [1734]. In particular, the polypeptide chains of normal prion and scrapie prion are identical in composition but differ in their 3-dimensional folded structures: normal prion is rich in alpha helices (spiral-like formations of amino acids) with few beta sheets (flattened strands of amino acids), whereas scrapie prion is poorer in alpha-helices and has many more beta-sheet domains [1733]. This structural transition from alpha-helices to beta-sheet in prion protein is apparently the fundamental event underlying prion disease [1733].* Since scrapie prions can induce ordinary prion protein to refold into more scrapie protein, they represent an example of a self-replicating molecule [1368, 1736-1738] that employs a self-replicating geometry roughly analogous to the Penrose blocks (Section 3.3). The fastest-reported prion refolding event measured to date [1739] is for mPrP(121-231), the ~15.7 kD structured 111-residue domain of the murine cellular prion protein PrPC (a strongly conserved cell surface glycoprotein totaling 231 amino acids in size), and occurs without kinetic intermediates at an extrapolated rate at 4 oC of 4000 sec-1 (~170 microsec half-life).
* The abnormal isoform also differs physically from the normal cellular isoform by its insolubility in detergents, its propensity to aggregate, and its relative resistance to breakdown by hydrolysis [1740]. Limited proteolysis of scrapie prion produces a smaller and protease-resistant molecule of approximately 142 amino acids which polymerizes into amyloid [1733]. Prions also appear to encode strain-specific properties in the tertiary structure of the modified prion protein [1734].
Prions are responsible for the transmissible spongiform encephalopathies (TSEs) including mad cow disease, scrapie, and the human Creutzfeldt-Jakob disease.
Last updated on 1 August 2005
{kind=link}