Kinematic Self-Replicating Machines
© 2004 Robert A. Freitas Jr. and Ralph C. Merkle. All Rights Reserved.
Robert A. Freitas Jr., Ralph C. Merkle, Kinematic Self-Replicating Machines, Landes Bioscience, Georgetown, TX, 2004.
4.1.1 Self-Assembling Peptides, Porphyrins, Nucleotides and DNA
One approach using solid-phase peptide synthesis and a convergent self assembly process was employed by M.R. Ghadiri and colleagues [1393-1398], who designed and synthesized a number of self-assembling peptide-based organic nanotubes using cyclic peptides with alternating D- and L-amino acids for the building blocks of the nanotubes. The alternating stereochemistry of the cyclic peptides allowed all the side chains of the amino acids to be pointing outwards which would not be possible in an ordinary all L-cyclic peptide. In this conformation, the amide backbone can H-bond in a direction perpendicular to the plane of the cyclic peptide. The stacking of two cyclic peptides forms an H-bonding network resembling an anti-parallel beta-sheet [1399] as is commonly found in natural proteins. The H-bonding lattice quickly propagates perpendicular to the plane of the cyclic peptide, forming a tubular microcrystalline structure with 0.75-nm pores, or, in another experiment, 1.3-nm pores [1400]. Another group of nanotubes was designed with a highly hydrophobic outer surface and a hydrophilic inner pore. These nanotubes were easily inserted into a lipid bilayer and have been shown to be highly efficient ion channels [1397, 1401]. Nanotubes with slightly larger pores transport small molecules such as glucose as well [1402]. Stable flat nanodisks with diameters continuously (chemically) adjustable from 30-3000 nm have been self-assembled in surfactant solutions [1403]. Other groups have investigated self-assembling peptides to form nanotubes and nanovesicles [1404-1406].
Yokoyama et al [1407] report the formation of surface-supported supramolecular structures whose size and spontaneous aggregation pattern are rationally controlled by tuning the selective and directional non-covalent interactions between individual substituted porphyrin molecule “molecular building blocks” adsorbed on a gold surface that form monomers, trimers, tetramers or extended wire-like structures. “We find that each structure corresponds in a predictable fashion to the geometric and chemical nature of the porphyrin substituents that mediate the interactions between individual adsorbed molecules,” note the researchers. “Our findings suggest that careful placement of functional groups that are able to participate in directed non-covalent interactions will allow the rational design and construction of a wide range of supramolecular architectures absorbed to surfaces, [potentially enabling] the realization of molecule-based miniature devices with advanced functions.” Crossley’s group [1408] is also studying self-replication and templated synthesis of porphyrin systems [1409].
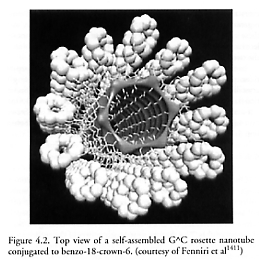
Fenniri et al [1410-1413] have used the heteroaromatic bicyclic base G^C which possesses the Watson-Crick donor-donor-acceptor of guanine (G) and acceptor-acceptor-donor of cytosine (C) at a fixed 60o relative angle, to achieve the self-assembly of helical rosette nanotubes which stack (six per hexagonal ring) only in the correct orientation because they are hydrophobic on the inside and hydrophilic on the outside (Figure 4.2). “Because of the disymmetry of its hydrogen bonding arrays, their spatial arrangement, and the hydrophobic character of the bicyclic system, G^C undergoes a hierarchical self-assembly process under physiological conditions to form a six-membered supermacrocycle maintained by 18 H bonds (rosette). The resulting and substantially more hydrophobic aggregate then undergoes a second level of organization to produce a stack. The architecture thus generated defines an unoccluded central pore running the length of the stack with tunable inner and outer diameters. The inner space is directly related to the distance separating the H-bonding arrays within the G^C motif, while the peripheral diameter and its chemistry are dictated by the choice of the functional groups conjugated to this motif.” Dendrimeric oligonucleotides as stable nanostructures have also been investigated by others [1414].
The ideas behind self-assembled DNA nanotechnology have been around since 1980 [1438], but activity in this field accelerated in the 1990s after numerous experimental difficulties were surmounted. Horn and Urdea [1415] reported branched and forked DNA polymers. Niemeyer [1416] and Smith [1417, 1452] suggested using self-assembled DNA as an early material for molecular nanotechnology. Damha [1418, 1419] synthesized V-shaped and Y-shaped branched RNA molecules, branching RNA dendrimers with “forked” and “lariat” shaped RNA intermediates [1420], and even trihelical DNA [1421]. Henderson and coworkers [1422-1425] designed a simple DNA decamer that can form an extended linear staggered quadruplex array reaching lengths of >1000 nm, and have made branched oligonucleotides that template the synthesis of their branched “G-wires”; depending on the ratio of linear to branched building blocks, extensive DNA arrays with differing connectivities but irregular interstices can be created [1426]. Von Kiedrowski’s group [1363, 1426-1428] has generated 3-D nanoobjects using trisoligonucleotides with covalent junctions whose self-assembly leads to noncovalent objects, which can then be selectively replicated using an electrophoretic process called eSPREAD [1427, 1428]. Reif’s group [1431] has produced self-assembled DNA-based nanotubes, and various self-assembled DNA-based dendrimers have been produced [1432, 1433].
Some of the most intensive and sustained work on three-dimensional engineered DNA structures has taken place in Nadrian Seeman’s laboratory [1440-1450] in the New York University Department of Chemistry. Seeman originally conceived the idea of rigid 3-D DNA structures in the early 1980s [1438, 1458] while examining DNA strands that had arranged themselves into unusual four-armed Holliday junctions [1459]. Seeman recognized that DNA had many advantages as a construction material for nanomechanical structures [1444]. First, each double-strand DNA with a single-strand overhang has a “sticky end,” so the intermolecular interaction between two strands with sticky ends is readily programmed (due to base-pair specificity) and reliably predicted, and the local structure at the interface is known (sticky ends associate to form B-DNA). Second, arbitrary sequences are readily manufactured using conventional biotechnological techniques. Third, DNA can be manipulated and modified by a large variety of enzymes, including DNA ligase, restriction endonucleases, kinases and exonucleases. Fourth, DNA is a stiff polymer in 1-3 turn lengths [1460] and has an external code that can be read by proteins and nucleic acids [1461].
During the 1980s, Seeman worked to develop strands of DNA that would zip themselves up into more and more complex shapes. Seeman made junctions with five and six arms, then squares [1462], stick-figure cubes comprised of 480 nucleotides [1440], and a truncated octahedron containing 2550 nucleotides and a molecular weight of ~790,000 daltons [1443]. The cubes were synthesized in solution, but Seeman switched to a solid-support-based methodology [1463] in 1992, greatly improving control by allowing construction of one edge at a time and isolating the growing objects from one another, allowing massively parallel self-assembly of objects with far greater control of the synthesis sequence. By the mid-1990s, most Platonic (tetrahedron, cube, octahedron, dodecahedron, and icosahedron), Archimedean (e.g. truncated Platonics, semiregular prisms and prismoids, cuboctahedron, etc.), Catalan (linked rings and complex knots), and irregular polyhedra could be constructed as nanoscale DNA stick figures [1141, 1151].
Seeman’s DNA strands that formed the frame figures were strong enough to serve as girders in a molecular framework, but the junctions were too floppy. In 1993 Seeman discovered the more rigid antiparallel DNA “double crossover” motif [1464], which in 1996 he used to design and build a stiff double junction to keep his structures from sagging [1465]. The next goal was to bring together a large number of stick figures to form large arrays of cage-shaped DNA crystals [1437] that could then be used as frameworks for the assembly of other molecules into pre-established patterns. These DNA molecules, including the newer “triple crossover” motifs [1446], would serve as the scaffolding upon which new materials having precise molecular structure could be assembled [1447-1449]. Seeman’s DNA nanoconstruction work has more recently began progressing toward DNA-based kinematic nanodevices (Section 4.5).
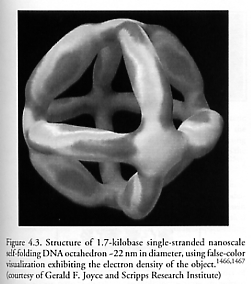
In 2004, Shih et al [1466, 1467] at the Scripps Research Institute synthesized a single-stranded 1669-nucleotide DNA molecule that self-folds into a hollow octahedral structure ~22 nm in diameter in the presence of five 40-nucleotide synthetic oligodeoxynucleotides by a simple denaturation-renaturation process involving heating and a series of cooling steps (Figure 4.3). The central cavity can hold a sphere 14 nm in diameter, which would include fullerenes up to ~C9,200 in size; the triangular openings on each face can admit spheres up to 8 nm (~C2,900) in outside diameter. The base-pair sequence of individual struts is not repeated in a given octahedron, so each strut is uniquely addressable by the appropriate sequence-specific DNA binder [1466]. Unlike other DNA that forms three-dimensional objects, Shih’s molecules can be readily copied by polymerases, hence is clonable. “Building complex three-dimensional DNA wireframes directly from synthetic oligonucleotides (as has been done in the past) is a very time-consuming and inefficient process, and thus not practical for many applications,” said Shih in a February 2004 interview [1467]. “Polymerases could also facilitate the discovery of novel single strand encoded DNA molecular machines through the procedure known as directed molecular evolution. Directed evolution could be used for attempts to develop complex mechanical behavior from DNA loops organized by an octahedron scaffold. This could lead to custom molecular machines reminiscent of those found in nature – for example, molecular assemblers and disassemblers such as chaperones or polymerase clamp loaders. Directed evolution has already been used successfully to discover static and even allosterically controlled catalysts. It would be a great feat to evolve more complicated molecular machines, and amplifiable DNA nano-scaffolds could play a key role in meeting this challenge.” Wengel [3278] has also discussed using DNA building blocks for nanoconstruction.
Last updated on 1 August 2005
{kind=link}
{kind=link}